Requisitos y procesos de certificación para exportar dispositivos médicos a Australia.
Category: Equipos MédicosDate: 31 de octubre de 2024 10:55Source: Agente Imp. & Exp. China
Home»Equipos Médicos» Requisitos y procesos de certificación para exportar dispositivos médicos a Australia.
Australia cuenta con un sistema de atención médica público y privado maduro y de gran escala, que...Equipos MédicosLa demanda también es muy alta. Sin embargo, para vender, importar o exportar dispositivos médicos en Australia, es necesario completar el proceso de registro y obtener el certificado de acceso de acuerdo con las regulaciones correspondientes. Este artículo detallará este proceso.
Ya en 1966, Australia aprobó la "Ley de Productos Terapéuticos" para regular los productos médicos. Actualmente, las principales leyes de regulación son la "Ley de Productos Terapéuticos" de 1989 y el "Reglamento de Dispositivos Médicos" promulgado en 2002. La implementación y supervisión de estas dos regulaciones están a cargo del TGA, una agencia subordinada del Departamento Federal de Salud y Cuidado de Ancianos de Australia. Por lo tanto, para vender oImportación y exportaciónEl fabricante de dispositivos médicos debe presentar una solicitud de acceso al mercado a la TGA.
II. Definición y clasificación de dispositivos médicos en Australia
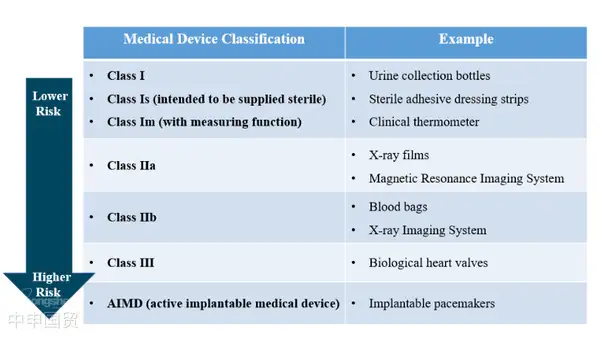
Australia define los dispositivos médicos como productos terapéuticos compuestos por herramientas, instrumentos, utensilios u otros artículos, junto con sus accesorios o software necesarios. Estos productos terapéuticos logran su función principal principalmente a través de medios no farmacológicos, inmunológicos o metabólicos, aunque estos métodos pueden complementar su efecto. Según el nivel de riesgo, los dispositivos médicos se clasifican en Clase I, Clase IIa, Clase IIb, Clase III y Clase AIMD (Dispositivos Médicos Implantables Activos).
III. Acceso al mercado de dispositivos médicos en Australia
La TGA clasifica los dispositivos médicos en tres categorías para su regulación: exentos, notificados y registrados. Cada tipo de dispositivo médico debe obtener la aprobación del gobierno antes de ser comercializado y cumplir con los requisitos básicos de los dispositivos médicos. Los dispositivos médicos de alto riesgo deben ser evaluados por la TGA y obtener aprobación antes de su lanzamiento al mercado. Mientras tanto, los dispositivos médicos de bajo riesgo pueden ser evaluados por las empresas por sí mismas, siempre que cumplan con las condiciones de calidad y seguridad para ingresar al mercado. Todos los dispositivos médicos fabricados en Australia deben cumplir con los estándares de GMP y ser producidos en un ambiente limpio y libre de contaminación.
IV. Análisis del proceso de comercialización de dispositivos médicos en Australia
El proceso de comercialización de dispositivos médicos en Australia es un trabajo riguroso y tedioso, que requiere cumplir con las responsabilidades de múltiples roles clave, definir claramente la clasificación del dispositivo médico y su nivel de riesgo, así como completar la evaluación de conformidad y el registro en el ARTG.
(1) Definir los roles clave En primer lugar, el proceso de registro y comercialización de dispositivos médicos en Australia involucra dos roles clave: el fabricante (Manufacturer) y el patrocinador (Sponsor).
El fabricante es responsable del dise?o, producción, empaquetado y envío del dispositivo médico, mientras que el patrocinador debe ser un ciudadano o empresa local de Australia. Su función es solicitar un número de registro de Sponsor ante la TGA (Administración de Productos Terapéuticos) para facilitar el proceso de registro.
(2) Definir claramente la definición y los niveles de riesgo de los dispositivos médicos. A continuación, es necesario definir claramente el concepto de dispositivo médico y sus niveles de riesgo. La "Ley de Productos Médicos" de 1989 establece la definición y los puntos clave de los dispositivos médicos de la siguiente manera:
En Australia, los dispositivos médicos que cumplen con la definición de la Ley de Productos Terapéuticos de 1989 deben presentar una solicitud de registro ante la TGA. Según el nivel de riesgo y el uso previsto, los dispositivos médicos se clasifican en cinco categorías de riesgo; cuanto mayor sea el nivel de riesgo, más estricta será la regulación de la TGA.
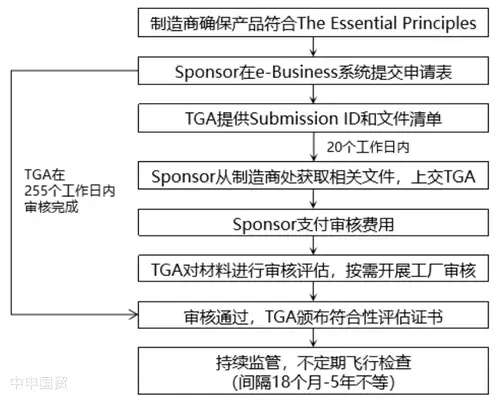
(3) Solicitud de certificado de evaluación de conformidad Antes del registro y comercialización de dispositivos médicos, los fabricantes deben implementar procedimientos de evaluación de conformidad calificados y someterse a la auditoría de la TGA para obtener un certificado de evaluación de conformidad válido.
Los certificados de evaluación de conformidad reconocidos por la TGA incluyen:
- TGA Conformity Assessment Evidence,
- EC Certificate,
- y los certificados emitidos bajo los acuerdos EC-MRA y EFTA-MRA. Si se elige la evidencia de evaluación de conformidad TGA, el proceso de solicitud del certificado es el siguiente:
(4) Presentar el registro ante el ARTG Después de completar la evaluación de conformidad, el fabricante y el patrocinador deben presentar el registro en el ARTG para obtener el certificado de ingreso de un dispositivo médico individual y así completar el listado del producto. Solo cuando el dispositivo médico esté incluido en el Registro de Productos Terapéuticos de Australia (ARTG), podrá ser comercializado en el mercado australiano.
V. Sistema de calidad de dispositivos médicos en Australia
El gobierno establece que todos los procesos de producción de las empresas fabricantes de dispositivos médicos deben cumplir con los requisitos de calidad relacionados con los dispositivos que producen, y deben contar con los medios y procedimientos de garantía de calidad. Para lograr este objetivo, Australia implementó el sistema de Buenas Prácticas de Manufactura (GMP) y también se alineó con los estándares del sistema de calidad ISO 9000. A partir de 2017, Australia comenzó a implementar el Programa de Auditoría única para Dispositivos Médicos (MDSAP) y acepta los certificados de MDSAP emitidos por organismos de inspección calificados.
VI. Gestión poscomercialización de productos de dispositivos médicos en Australia
Esto se logra principalmente a través del sistema de gestión de vigilancia poscomercialización. Australia garantiza que todos los dispositivos médicos en el mercado cumplan con las regulaciones mediante informes de investigación de eventos adversos, pruebas de laboratorio y actividades de monitoreo de productos comercializados. Sus regulaciones de vigilancia de eventos adversos poscomercialización son muy maduras, con procedimientos detallados que combinan principios y flexibilidad, lo que las hace altamente operativas.
VII. Regulaciones sobre modificaciones de registro de productos de dispositivos médicos en Australia.
Después de que un producto obtiene la autorización para su comercialización, su información y estado deben seguir siendo supervisados continuamente. Si el producto experimenta cambios sustanciales, el fabricante y el patrocinador deben notificar oportunamente a la Administración de Productos Terapéuticos (TGA). El patrocinador debe iniciar sesión en el sistema de e-Business para presentar una solicitud de modificación de registro. Estos cambios pueden incluir modificaciones en la información del fabricante, patrocinador o del producto, cambios en la información del certificado de evaluación de conformidad, alteraciones en el uso previsto del producto o sus indicaciones clínicas, así como cambios en la categoría del producto o su código GMDN.
En resumen, para vender, importar o exportar dispositivos médicos en Australia, es necesario pasar por una serie de procesos estrictos de admisión y registro. Estos procesos garantizan la calidad, seguridad y eficacia de los dispositivos médicos, lo que a su vez protege la calidad del sistema de atención médica.